-
+90 530 762 72 18
Please feel free to contact our friendly reception staff with any general or medical enquiry.
Create an Appointment - Confidence in Healthcare








Phenylketonuria: Causes, Symptoms, and Diagnosis
Phenylketonuria (PKU) is a genetic disease that causes the amino acid called phenylalanine to accumulate in the body. Phenylalanine is found in all proteins and some of the artificial sweeteners. Early diagnosis and treatment in this disease are necessary to alleviate symptoms and prevent brain damage. For this reason, the blood phenylalanine levels should be measured twice a week in the first 2 years of age, once a week at the age of 2 to 4 years, once every 15 days at the age of 4 to 10 years, and once a month in the following periods. These levels are expected to be 2-6 mg/dl in the period between 0-12 years and 2-11 mg/dl in the period of 12 years and beyond. During pregnancy of women with PKU, this value should be kept between 2-4 mg/dl.
What is Phenylketonuria?
“What is phenylketonuria disease?” The answer can be given as an inherited disease that increases the level of a substance called phenylalanine in the blood. Phenylalanine, which is the building block of dietary proteins, is found in all proteins and in some artificial sweeteners. If this disease, also known as PKU, is not treated, phenylalanine can reach harmful levels in the body and cause many serious health problems or mental disability.
Who Gets Phenylketonuria?
“What causes phenylketonuria and who gets it?” If you are wondering, you should know that this disease is seen in people who carry a homozygous mutation in the gene encoding the phenylalanine enzyme. Parents who have this gene transmit the disease to their offspring, and this inheritance pattern is called autosomal recessive. In addition, if only one of the parents has the defective gene, there is no risk of passing the disease to the child, but the child may carry the risk of being a carrier.
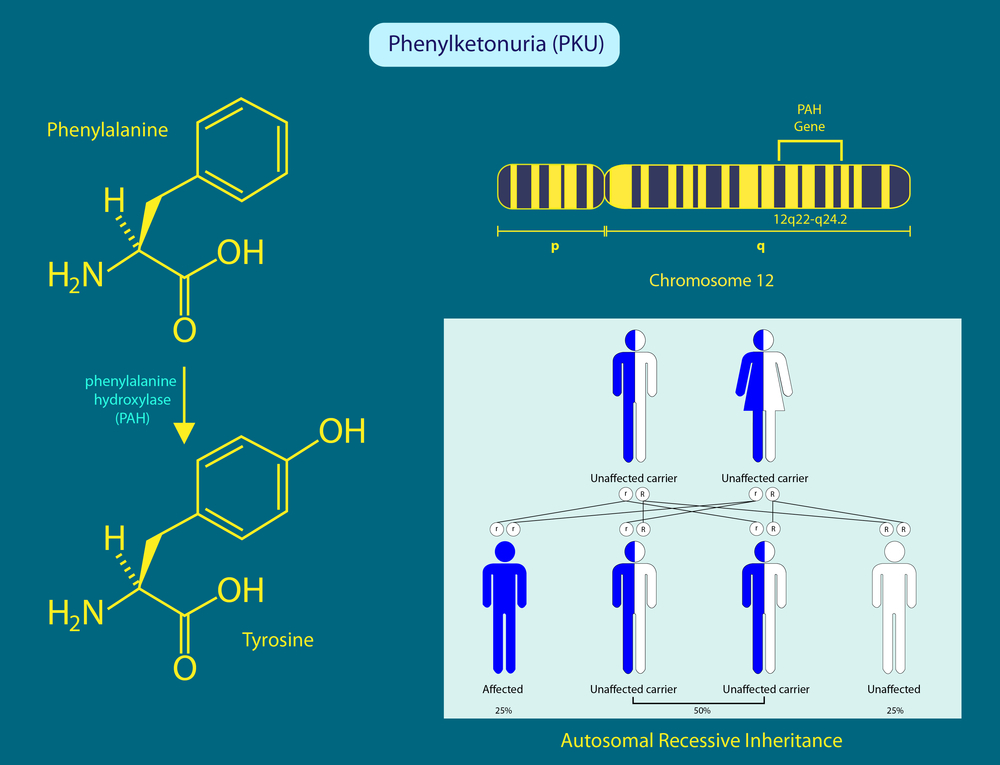
What Causes Phenylketonuria?
“What is phenylketonuria? What are the reasons?” The question can be answered as this ailment occurs due to a disorder in the metabolism of phenylalanine. Phenylalanine is an essential amino acid for the body and must be obtained from food. In normal condition, phenylalanine is converted to tyrosine by its enzyme in the body. However, if there is an error in this cycle, the amino acid in protein foods cannot be broken down in the liver, and its level in the blood increases. With this increase, accumulation is seen in the blood, tissues and brain, and as a result, phenylketonuria is experienced.
Phenylketonuria (PKU) Test
The phenylketonuria test provides enough data to diagnose the majority of cases. However, people who have PKU or have a family history of PKU should have prenatal screening tests. The screening test is done with blood taken from the heel when the baby is 1 to 2 days old. This test shows whether the baby has the disease. However, in some cases, more urine and blood tests may be required to strengthen the Phenylketonuria diagnosis.
What are the Symptoms of Phenylketonuria?
No symptoms are observed in babies born with this disease. Failure to make the necessary diagnosis causes the baby to have difficulty in diapering, vomiting and skin rashes in the following months. If the treatment is not started, excessive accumulation of phenylalanine in the brain may cause neurological findings and mental problems. Babies with this disease usually have an IQ below 50. These babies also have light skin, hair and eye color. Phenylketonuria symptoms are generally as follows:
- Fair skin and blue eyes
- Mental retardation
- Psychological problems
- Neurological problems and seizures
- Hyperactivity
- Eczema
- Musty smell of breath, skin or urine
- Abnormally smaller than normal head size
- Behavioural problems
- Emotional and social problems
What are the Physical Characteristics of Phenylketonuria Patients?
“What does phenylketonuria mean?” As well as the question, the physical characteristics of people who have this disorder are also a matter of curiosity. The growth and development of children with this disorder lag behind their peers. Their heads are also small because the brain is poorly developed. Children with PKU also have pigmentation disorders, so the child's skin color is noticeably lighter than their parents' and their eyes are blue.

How is Phenylketonuria Diagnosed?
Treatment of phenylketonuria depends on the correct diagnosis. PKU, which is an inherited condition, must be diagnosed in infancy. For this reason, it is one of the important neonatal screenings performed for newborn babies. The diagnosis can be made by blood taken from the heel of the newborn baby. This blood is taken from each newborn baby within 24 to 48 hours. In order to prevent the test from giving false results, the blood sample is taken after the first 24 hours following the baby's adequate protein intake. However, in babies who are discharged before the first 24 hours, the blood sample is taken again within the first week. The level of phenylalanine in the blood of babies with a positive test result is checked. The expected value is 120-360 micromol/l. A value above 360 micromol/l (6 mg/dl) indicates PKU, and 600 micromol/l (10 mg/dl) indicates classical PKU. In addition, a new evaluation is made by measuring the phenylalanine levels in the blood at the 2nd and 4th weeks after birth for babies with a negative screening test but a family history of PKU.
How is the Treatment of Phenylketonuria Disease?
Phenylketonuria treatment is to follow a restricted diet from birth. However, early diagnosis is important for treatment. Most of the patients have normal intelligence after PKU treatment if the diagnosis is made in the first 10-20 days. However, it is important that the treatment continues for life in order to prevent physical and neurological deterioration of the person. During the treatment process, the plasma level of phenylalanine is kept between 120-360 micromol/l (2-6 mg/dl).
Who Should Get Phenylketonuria Treatment?
The treatment is started as soon as the symptoms of phenylketonuria are observed, even right after birth. Within this process:
Newborn babies are screened. If PKU is detected in the baby, the necessary dietary modifications are made.
Expectant mothers also go through the treatment process. Women who have a family history of PKU and are in childbearing age should follow a phenylketonuria diet planned by a specialist before and during pregnancy.
Can Phenylketonuria Patients Be Fed with Breast Milk?
Phenylketonuria is not a condition that prevents breastfeeding. However, in this process, medical formulas and blood values for the baby should be followed closely.
Is Disease Prevention Possible?
Since this disease is an inherited condition, it is not possible to prevent it, but a healthy life can be achieved with the diet of phenylketonuria disease.
Can a Mother with a Diagnosis of Phenylketonuria Have a Child?
If the expectant mother has received regular treatment throughout her life and continued the treatment during pregnancy, the baby may come to the world healthy, but the baby will be a carrier.

Phenylketonuria Diet
Nutrition is of great importance in the treatment process of phenylketonuria. The products that patients should not consume are as follows:
- Milk and dairy products, all foods made from these products
- Meat products
- Bread
- Egg
- Internal organs of animals
- Hazelnut, walnut, pistachio, kernel varieties, roasted chickpeas, almonds
- Dry beans
- Prepared foods
- All foods and beverages containing aspartame and phenylalanine
The diet of these patients is maintained throughout life. For this reason, the diet of babies with PKU should be established from the first period of their lives and should be followed by a specialist. The food list given by the expert; This makes it necessary to completely avoid foods high in protein, such as eggs, dairy products, and meat. The patient’s consumption of foods such as potatoes and cereals are also controlled.






