-
+90 530 762 72 18
Bei allgemeinen oder medizinischen Fragen wenden Sie sich bitte an unser freundliches Personal an der Rezeption.
Erstellen Sie einen Termin - Reliable Healthcare 28th years








Phenylketonurie: Symptome, Vererbung, Therapie
Phenylketonurie (PKU) ist eine genetisch bedingte Störung, bei der sich eine Mineralsäure namens Phenylalanin im Körper ansammelt. Phenylalanin ist in allen Proteinen und einigen künstlichen Süßungsmitteln enthalten. Bei dieser Krankheit ist eine frühzeitige Diagnose und Behandlung notwendig, um die Symptome zu lindern und Hirnschäden zu verhindern. Aus diesem Grund sollte der Phenylalaninspiegel im Blut des Kindes in den ersten zwei Lebensjahren zweimal wöchentlich, zwischen dem zweiten und vierten Lebensjahr einmal wöchentlich, zwischen dem vierten und zehnten Lebensjahr einmal alle 15 Tage und in den folgenden Lebensjahren einmal im Monat gemessen werden. Es wird erwartet, dass diese Werte zwischen 0 und 12 Jahren bei 2-6 mg/dl und nach 12 Jahren bei 2-11 mg/dl liegen. Während der Schwangerschaft von Frauen mit PKU-Erkrankung sollte dieser Wert zwischen 2-4 mg/dl gehalten werden.
Was ist Phenylketonurie?
Die Frage "Was ist die Phenylketonurie-Krankheit?" kann als eine Erbkrankheit beantwortet werden, bei der der Gehalt an einer Substanz namens Phenylalanin im Blut erhöht ist. Phenylalanin, der Baustein der aus der Nahrung gewonnenen Proteine, ist in allen Proteinen und einigen künstlichen Süßstoffen enthalten. Wird diese Krankheit, die auch als PKU bezeichnet wird, nicht behandelt, kann der Phenylalaninspiegel im Körper auf ein schädliches Niveau ansteigen und viele schwere gesundheitliche Probleme oder geistige Behinderungen verursachen.
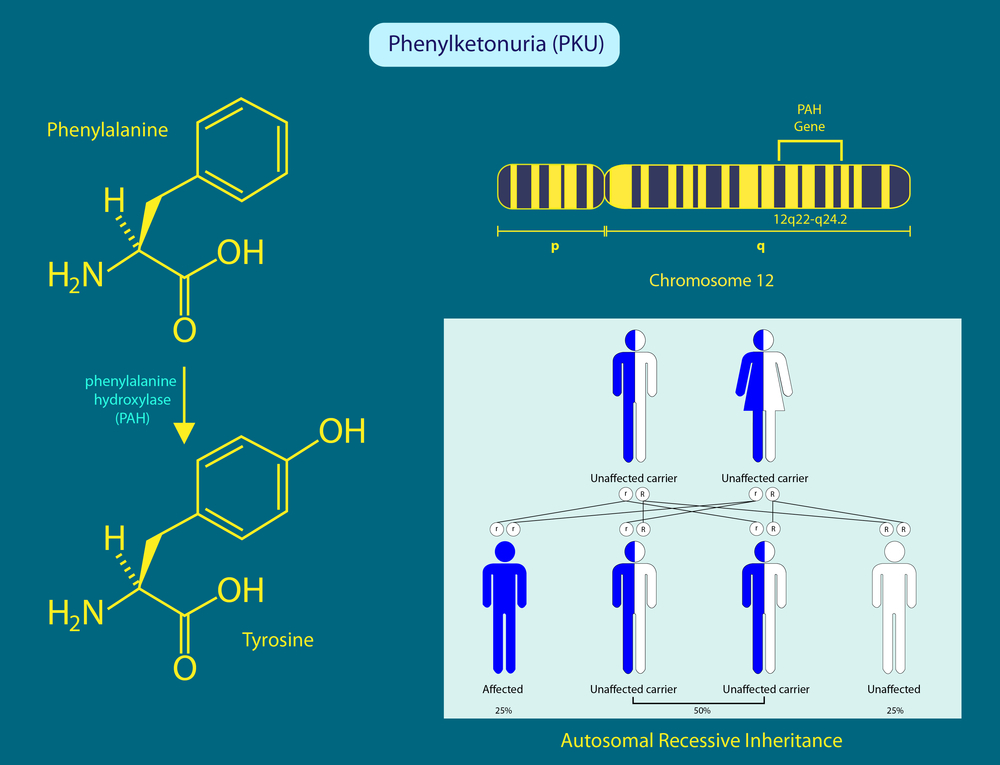
Bei wem liegt eine Phenylketonurie vor?
Wenn Sie sich fragen: "Was verursacht Phenylketonurie und wer hat sie?", sollten Sie wissen, dass diese Krankheit bei Menschen auftritt, die eine homozygote Mutation im Gen für das Enzym Phenylalanin tragen. Die Mutter und der Vater, die dieses Gen haben, geben die Krankheit an ihre Kinder weiter, und dieses Vererbungsmodell wird als autosomal rezessiv bezeichnet. Wenn nur ein Elternteil das defekte Gen hat, besteht kein Risiko, die Krankheit an das Kind weiterzugeben, doch das Kind kann ein Träger sein.
Wodurch wird Phenylketonurie verursacht?
"Was ist Phenylketonurie? Die Frage kann beantwortet werden, indem diese Störung auf eine Störung des Phenylalaninstoffwechsels zurückgeführt wird. Phenylalanin ist eine essentielle Aminosäure für den Körper und muss mit der Nahrung aufgenommen werden. Im Normalzustand wird Phenylalanin im Körper mit Hilfe eines Enzyms in Tyrosin umgewandelt und erfüllt diese Funktion. Liegt jedoch ein Fehler bei dieser Umwandlung vor, kann die Aminosäure in eiweißhaltigen Lebensmitteln in der Leber nicht abgebaut werden, und ihr Gehalt im Blut steigt an. Bei diesem Anstieg kommt es zu einer Anreicherung im Blut, im Gewebe und im Gehirn, was zu Phenylketonurie führt.
Test auf Phenylketonurie (PKU)
Der Phenylketonurie-Test liefert ausreichende Daten, um die meisten Fälle zu diagnostizieren. Jedoch sollten Menschen mit PKU oder einer familiären Vorbelastung mit PKU vor der Schwangerschaft oder Geburt Vorsorgeuntersuchungen durchführen lassen. Der Screening-Test wird mit einer Blutentnahme aus der Ferse durchgeführt, wenn das Baby 1 bis 2 Tage alt ist. Dieser Test zeigt, ob das Baby die Krankheit hat. In einigen Fällen können jedoch weitere Urin- und Bluttests erforderlich sein, um die Diagnose zu sichern.
Was sind die Symptome der Phenylketonurie?
Phenylketonurie ist eine Krankheit mit Symptomen. Bei Säuglingen, die mit dieser Krankheit geboren werden, werden jedoch keine Symptome beobachtet. Wenn die erforderliche Diagnose nicht durchgeführt wird, leidet das Baby in den folgenden Monaten unter Schwierigkeiten beim Wickeln, Erbrechen und Hautausschlägen. Wenn nach diesen Diagnosen keine Behandlung eingeleitet wird, kann Phenylalanin durch übermäßige Anreicherung im Gehirn neurologische Befunde und geistige Probleme verursachen. Der IQ von Säuglingen mit dieser Krankheit liegt in der Regel unter 50. Diese Babys haben auch eine helle Haut-, Haar- und Augenfarbe. Die Symptome der Phenylketonurie-Krankheit sind im Allgemeinen wie folgt:
- Helle Hautfarbe und blaue Augen
- Mentale Retardierung
- Psychologische Probleme
- Neurologische Probleme und Krampfanfälle
- Hyperaktivität
- Ekzem
- Schimmeliger Geruch von Atem, Haut oder Urin
- Abnorm kleiner als der normale Kopfumfang
- Verhaltensauffälligkeiten
- Emotionale und soziale Probleme
Was sind die körperlichen Merkmale von Phenylketonurie-Patienten?
"Auch die Frage "Was bedeutet Phenylketonurie?" und die körperlichen Merkmale von Menschen mit dieser Krankheit sind von Interesse. Die Entwicklung und das Wachstum von Kindern mit dieser Störung bleiben hinter denen ihrer Gleichaltrigen zurück. Da das Gehirn unterentwickelt ist, ist auch ihr Kopf klein. Außerdem haben Kinder mit PKU eine Pigmentierungsstörung, weshalb ihre Hautfarbe deutlich heller ist und ihre Augen blau sind als die ihrer Eltern.

Wie wird Phenylketonurie diagnostiziert?
Die Behandlung der Phenylketonurie-Erkrankung hängt von der richtigen Diagnose ab. PKU ist eine Erbkrankheit, die bereits im Säuglingsalter diagnostiziert werden sollte. Deshalb gehört sie zu den wichtigen Vorsorgeuntersuchungen für Neugeborene. Die Diagnose kann durch eine Blutentnahme an der Ferse des Neugeborenen gestellt werden. Dieses Blut wird jedem Neugeborenen innerhalb von 24 bis 48 Stunden entnommen. Um falsche Ergebnisse zu vermeiden, wird die Blutprobe nach den ersten 24 Stunden nach einer ausreichenden Eiweißzufuhr des Babys entnommen. Bei Säuglingen, die vor Ablauf der ersten 24 Stunden entlassen werden, wird die Blutprobe jedoch innerhalb der ersten Woche erneut entnommen. Bei Säuglingen mit einem positiven Testergebnis wird der Phenylalaninspiegel im Blut analysiert. Der erwartete Wert für diesen liegt bei 120-360 Mikromol/l. Ein Wert von über 360 Mikromol/l (6 mg/dl) weist auf PKU hin, ein Wert von 600 Mikromol/l (10 mg/dl) auf klassische PKU. Darüber hinaus wird bei Säuglingen, deren Screening-Test negativ ausfällt, bei denen aber PKU in der Familienanamnese vorkommt, eine erneute Untersuchung durchgeführt, bei der die Phenylalaninwerte im Blut in der zweiten und vierten Woche nach der Geburt kontrolliert werden.
Wie wird die Phenylketonurie behandelt?
Die Behandlung der Phenylketonurie besteht darin, dem Kind von Geburt an eine beschränkte Diät zu verabreichen. Wichtig für die Behandlung ist jedoch eine frühzeitige Diagnose. Bei den meisten Patienten ist die Intelligenz nach einer PKU-Behandlung, die innerhalb der ersten 10-20 Tage diagnostiziert wird, normal. Es ist jedoch wichtig, die Behandlung lebenslang fortzusetzen, um körperliche und neurologische Verschlechterungen zu verhindern. Während der Behandlung wird der Plasmaspiegel zwischen 120-360 Mikromol/l (2-6 mg/dl) gehalten.
Wer sollte eine Phenylketonurie-Behandlung erhalten?
Der Behandlungsprozess beginnt mit der Diagnose der Phenylketonurie ab dem Zeitpunkt, an dem die Symptome auftreten, oder sogar unmittelbar nach der Geburt. Im Rahmen dieses Prozesses:
Screening-Tests werden bei Neugeborenen durchgeführt. Wird bei dem Kind durch den Test PKU festgestellt, wird die notwendige Ernährungstherapie eingeleitet.
Auch werdende Mütter durchlaufen einen Behandlungsprozess. Frauen, in deren Familie PKU vorkommt und die sich in der fruchtbaren Phase befinden, sollten vor und während der Schwangerschaft eine von einem Spezialisten geplante Phenylketonurie-Diät einhalten.
Können Phenylketonurie-Patienten gestillt werden?
Phenylketonurie ist kein Hindernis für das Stillen. In diesem Prozess sollten jedoch medizinische Formeln und Blutwerte des Babys genau beobachtet werden.
Ist es möglich, die Krankheit zu verhindern?
Da diese Erkrankung vererbt wird, ist es nicht möglich, sie zu verhindern, aber man kann mit einer Phenylketonurie-Diät ein gesundes Leben führen.
Kann eine Mutter mit Phenylketonurie ein Kind bekommen?
Wenn die werdende Mutter, die Symptome einer Phenylketonurie aufweist, ihr Leben lang regelmäßig behandelt wurde und die Behandlung während der Schwangerschaft fortsetzt, kann das Kind gesund zur Welt kommen, allerdings wird das Kind ein Träger sein.

Phenylketonurie-Diät
Bei der Phenylketonurie-Erkrankung ist die Ernährung von großer Bedeutung für die Behandlung. Die Produkte, die die Patienten in diesem Prozess nicht konsumieren sollten, sind folgende
- Milch und Molkereiprodukte, alle aus diesen Produkten hergestellten Lebensmittel
- Fleisch und Fleischerzeugnisse
- Normales Brot
- Ei
- Innere Organe des Tieres
- Haselnuss, Walnuss, Pistazie, Kernsorten, Kichererbsen, Mandeln
- Getrocknete Hülsenfrüchte
- Zubereitete Lebensmittel
- Alle Lebensmittel und Getränke, die Aspartam und Phenylalanin enthalten
Die Ernährung dieser Patienten wird ein Leben lang beibehalten. Aus diesem Grund sollte die Ernährung von Säuglingen mit PKU-Krankheit bereits in den ersten Lebensjahren festgelegt und von einem Spezialisten überwacht werden. Die vom Facharzt vorgegebene Nahrungsmittelliste erfordert den vollständigen Verzicht auf Lebensmittel mit hohem Eiweißgehalt wie Eier, Milchprodukte und Fleisch. Dabei wird auch der Verzehr von Lebensmitteln wie Kartoffeln und Getreide kontrolliert.






